Kurs
Unüberwachtes Lernen in R
4 Std.
55.1K
Es gibt ein paar Dinge, auf die du achten solltest, bevor du anfängst.
Es ist unbedingt notwendig, dass du die Skala deiner Merkmalswerte normalisierst, um mit dem Clustering-Prozess zu beginnen. Das liegt daran, dass die Merkmalswerte der einzelnen Beobachtungen als Koordinaten im n-dimensionalen Raum dargestellt werden (n ist die Anzahl der Merkmale) und dann die Abstände zwischen diesen Koordinaten berechnet werden. Wenn diese Koordinaten nicht normalisiert sind, kann das zu falschen Ergebnissen führen.
Nehmen wir zum Beispiel an, du hast Daten über Größe und Gewicht von drei Personen: A (1,75 m, 75 kg), B (1,77 m, 77 kg), C (1,75 m, 75 kg). Wenn du diese Merkmale in einem zweidimensionalen Koordinatensystem darstellst, Größe und Gewicht, und den euklidischen Abstand zwischen ihnen berechnest, wäre der Abstand zwischen den folgenden Paaren:
A-B : 2 Einheiten
A-C : 2 Einheiten
Nun, die Abstandsmetrik sagt, dass die beiden Paare A-B und A-C ähnlich sind, aber in Wirklichkeit sind sie es eindeutig nicht! Das Paar A-B ist ähnlicher als das Paar A-C. Daher ist es wichtig, diese Werte zuerst zu skalieren und dann den Abstand zu berechnen.
Es gibt verschiedene Möglichkeiten, die Merkmalswerte zu normalisieren. Du kannst entweder die gesamte Skala aller Merkmalswerte (x(i)) zwischen [0,1] standardisieren (bekannt als Min-Max-Normalisierung), indem du die folgende Transformation anwendest:
$ x(s) = x(i) - min(x)/(max(x) - min (x)) $
Du kannst die R-Funktion normalize() Funktion von R verwenden oder eine eigene Funktion schreiben, z. B:
standardize <- function(x){(x-min(x))/(max(x)-min(x))}
Eine andere Art der Skalierung kann durch die folgende Transformation erreicht werden:
$ x(s) = x(i)-mean(x) / sd(x) $
Dabei ist sd(x) die Standardabweichung der Merkmalswerte. So stellst du sicher, dass deine Verteilung der Merkmalswerte einen Mittelwert von 0 und eine Standardabweichung von 1 hat. Dies kannst du über die scale() Funktion in R.
Es ist auch wichtig, dass du dich vorher mit fehlenden/nullen/inf Werten in deinem Datensatz beschäftigst. Es gibt viele Möglichkeiten, mit solchen Werten umzugehen. Eine davon ist, sie entweder zu entfernen oder sie mit Mittelwert, Median, Modus zu imputieren oder einige fortgeschrittene Regressionstechniken anzuwenden. R hat viele Pakete und Funktionen, um mit Imputationen fehlender Werte umzugehen, wie impute(), Amelia, Mice, Hmisc usw. Du kannst über Amelia in diesem Tutorial lesen.
Die wichtigste Operation beim hierarchischen agglomerativen Clustering besteht darin, die beiden nächstgelegenen Cluster wiederholt zu einem größeren Cluster zusammenzufassen. Es gibt drei wichtige Fragen, die zuerst beantwortet werden müssen:
Wir hoffen, dass du am Ende dieses Tutorials in der Lage bist, alle diese Fragen zu beantworten. Bevor wir das hierarchische Clustering anwenden, schauen wir uns an, wie es funktioniert:
Es gibt verschiedene Möglichkeiten, den Abstand zwischen den Clustern zu messen, um die Regeln für das Clustering zu bestimmen, und sie werden oft Linkage-Methoden genannt. Einige der gängigen Verknüpfungsmethoden sind:
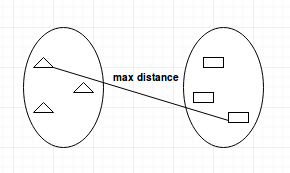
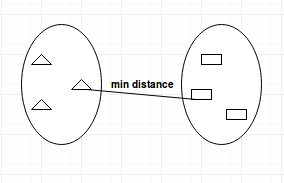
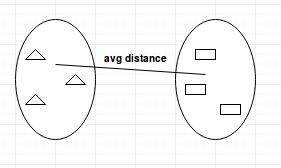
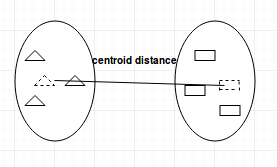
Die Wahl der Verknüpfungsmethode hängt ganz von dir ab und es gibt keine feste Methode, mit der du immer gute Ergebnisse erzielen kannst. Verschiedene Verknüpfungsmethoden führen zu unterschiedlichen Clustern.
Beim hierarchischen Clustering ordnest du die Objekte in eine Hierarchie ein, die einem baumartigen Diagramm ähnelt, das Dendrogramm genannt wird. Der Abstand der Aufspaltung oder Zusammenführung (Höhe genannt) wird auf der y-Achse des Dendrogramms unten angezeigt.
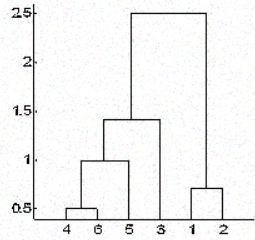
In der obigen Abbildung werden zunächst 4 und 6 zu einem Cluster, dem Cluster 1, zusammengefasst, da sie vom Abstand her am nächsten waren, gefolgt von den Punkten 1 und 2, dem Cluster 2. Danach wurde 5 im gleichen Cluster 1 zusammengeführt, gefolgt von 3, was zu zwei Clustern führte. Zuletzt werden die beiden Cluster zu einem einzigen Cluster zusammengeführt, und an dieser Stelle endet der Clustering-Prozess.
Eine Frage, die du dir vielleicht schon gestellt hast, ist, wie du entscheidest, wann du aufhörst, die Cluster zusammenzulegen? Das hängt davon ab, wie viel Wissen du über die Daten hast. Wenn du zum Beispiel Fußballspieler auf einem Feld anhand ihrer Positionen auf dem Feld clusterst, die ihre Koordinaten für die Entfernungsberechnung darstellen, weißt du bereits, dass du am Ende nur 2 Cluster haben solltest, da es nur zwei Mannschaften geben kann, die ein Fußballspiel spielen.
Aber manchmal hast du diese Informationen auch nicht. In solchen Fällen kannst du die Ergebnisse des Dendrogramms nutzen, um die Anzahl der Cluster zu ermitteln. Du schneidest den Dendrogrammbaum mit einer horizontalen Linie in einer Höhe, in der die Linie die maximale Strecke nach oben und unten zurücklegen kann, ohne den Zusammenführungspunkt zu schneiden. Im obigen Fall würde sie zwischen den Höhen 1,5 und 2,5 liegen, wie gezeigt:
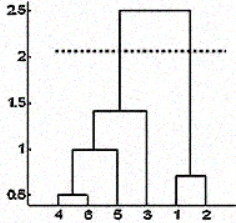
Wenn du den Schnitt wie gezeigt machst, hast du am Ende nur zwei Cluster.
Beachte, dass es nicht notwendig ist, nur an diesen Stellen einen Schnitt zu machen. Du kannst jeden beliebigen Punkt als Schnittpunkt wählen, je nachdem, wie viele Cluster du haben möchtest. Wenn du zum Beispiel unter 1,5 und über 1 schneidest, erhältst du 3 Cluster.
Beachte, dass dies keine feste Regel ist, um die Anzahl der Cluster zu bestimmen. Du kannst auch Diagramme wie das Silhouetten-Diagramm, das Ellbogen-Diagramm oder einige numerische Maße wie den Dunn-Index, das Hubert-Gamma usw. in Betracht ziehen, die die Variation des Fehlers mit der Anzahl der Cluster (k) zeigen, und du wählst den Wert von k, bei dem der Fehler am kleinsten ist.
Der vielleicht wichtigste Teil bei jeder unüberwachten Lernaufgabe ist die Analyse der Ergebnisse. Nachdem du das Clustering mit einem beliebigen Algorithmus und einer beliebigen Anzahl von Parametern durchgeführt hast, musst du sicherstellen, dass du es richtig gemacht hast. Aber wie kann man das feststellen?
Nun, es gibt viele Maßnahmen, um dies zu tun, die vielleicht beliebteste ist die Dunn's Index. Der Dunn-Index ist das Verhältnis zwischen den minimalen Inter-Cluster-Distanzen und dem maximalen Intra-Cluster-Durchmesser. Der Durchmesser eines Clusters ist der Abstand zwischen den beiden am weitesten entfernten Punkten. Um gut getrennte und kompakte Cluster zu erhalten, solltest du einen höheren Dunn-Index anstreben.
Jetzt wendest du das erworbene Wissen an, um ein Problem aus der realen Welt zu lösen.
Du wendest hierarchisches Clustering auf den Datensatz seeds an. Dieser Datensatz besteht aus Messungen geometrischer Eigenschaften von Körnern, die zu drei verschiedenen Weizensorten gehören: Kama, Rosa und Canadian. Sie enthält Variablen, die die Eigenschaften der Samen beschreiben, wie Fläche, Umfang, Asymmetriekoeffizient usw. Für jede Weizensorte gibt es 70 Beobachtungen. Die Details zum Datensatz findest du hier.
Beginne damit, den Datensatz mit der Funktion read.csv() in einen DataFrame zu importieren.
Beachte, dass die Datei keine Kopfzeilen hat und durch Tabulatoren getrennt ist. Um die Reproduzierbarkeit der Ergebnisse zu gewährleisten, musst du die Funktion set.seed() verwenden.
set.seed(786)
file_loc <- 'seeds.txt'
seeds_df <- read.csv(file_loc,sep = '\t',header = FALSE)Da der Datensatz keine Spaltennamen hat, gibst du den Spaltennamen selbst aus der Datenbeschreibung.
feature_name <- c('area','perimeter','compactness','length.of.kernel','width.of.kernal','asymmetry.coefficient','length.of.kernel.groove','type.of.seed')
colnames(seeds_df) <- feature_nameEs ist ratsam, einige grundlegende Informationen über den Datensatz zu sammeln, wie z. B. seine Dimensionen, Datentypen und -verteilung, Anzahl der NAs usw. Dazu verwendest du die Funktionen str(), summary() und is.na() in R.
str(seeds_df)
summary(seeds_df)
any(is.na(seeds_df))Beachte, dass dieser Datensatz alle Spalten als numerische Werte enthält. In diesem Datensatz gibt es keine fehlenden Werte, die du vor dem Clustering bereinigen musst. Aber die Skalen der Merkmale sind unterschiedlich und du musst sie normalisieren. Außerdem sind die Daten beschriftet und du hast bereits die Information, welche Beobachtung zu welcher Weizensorte gehört.
Jetzt speicherst du die Labels in einer separaten Variable und schließt die Spalte type.of.seed aus deinem Datensatz aus, um ein Clustering durchzuführen. Später kannst du anhand der echten Labels überprüfen, wie gut dein Clustering ausgefallen ist.
seeds_label <- seeds_df$type.of.seed
seeds_df$type.of.seed <- NULL
str(seeds_df)Wie du siehst, hast du die Spalte "True Label" aus deinem Datensatz entfernt.
Jetzt verwendest du die Funktion scale() von R, um alle deine Spaltenwerte zu skalieren.
seeds_df_sc <- as.data.frame(scale(seeds_df))
summary(seeds_df_sc)Beachte, dass der Mittelwert aller Spalten 0 ist und die Standardabweichung 1 beträgt. Jetzt, wo du deine Daten vorverarbeitet hast, ist es an der Zeit, die Distanzmatrix zu erstellen. Da alle Werte hier kontinuierliche Zahlenwerte sind, verwendest du die Methode euclidean distance.
dist_mat <- dist(seeds_df_sc, method = 'euclidean')An diesem Punkt solltest du dich entscheiden, welche Verknüpfungsmethode du verwenden möchtest und mit dem hierarchischen Clustering fortfahren. Du kannst alle möglichen Verknüpfungsmethoden ausprobieren und später entscheiden, welche davon besser funktioniert. Hier gehst du mit der Methode average linkage vor.
Du erstellst dein Dendrogramm, indem du das hierarchische Clusterobjekt zeichnest, das du mit hclust() erstellst. Du kannst die Verknüpfungsmethode über das Argument method angeben.
hclust_avg <- hclust(dist_mat, method = 'average')
plot(hclust_avg)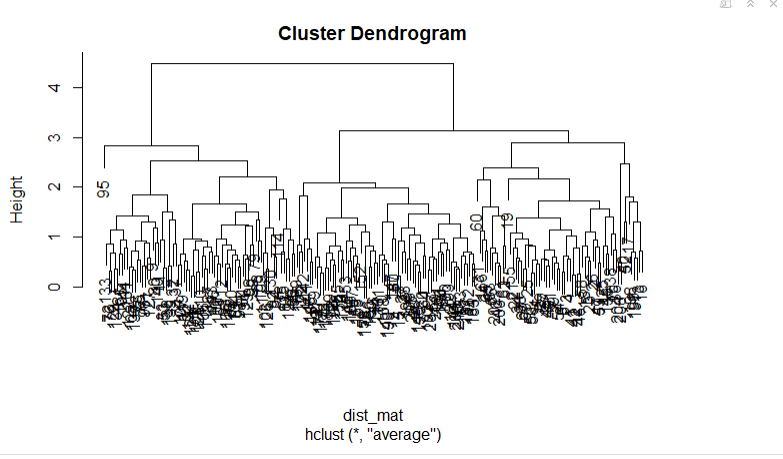
Beachte, wie das Dendrogramm aufgebaut ist und jeder Datenpunkt schließlich zu einem einzigen Cluster verschmilzt, wobei die Höhe (Entfernung) auf der y-Achse angezeigt wird.
Als Nächstes kannst du das Dendrogramm schneiden, um die gewünschte Anzahl von Clustern zu erstellen. Da du in diesem Fall bereits weißt, dass es nur drei Weizensorten geben kann, wählst du die Anzahl der Cluster k = 3, oder wie du im Dendrogramm h = 3 sehen kannst, erhältst du drei Cluster. Du wirst die Funktion cutree() von R verwenden, um den Baum mit hclust_avg als einem Parameter und dem anderen Parameter h = 3 oder k = 3 zu schneiden.
cut_avg <- cutree(hclust_avg, k = 3)Wenn du die Cluster im Dendrogramm visuell darstellen möchtest, kannst du die Funktion R's abline() verwenden, um die Schnittlinie zu zeichnen und rechteckige Fächer für jeden Cluster mit der Funktion rect.hclust() auf dem Baum zu überlagern, wie im folgenden Code gezeigt:
plot(hclust_avg)
rect.hclust(hclust_avg , k = 3, border = 2:6)
abline(h = 3, col = 'red')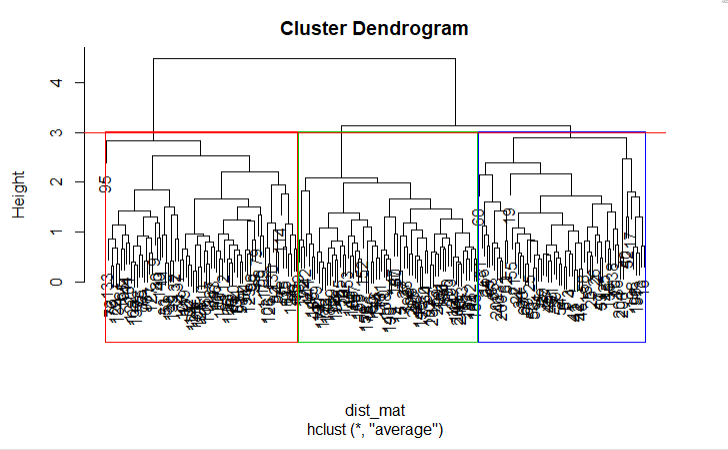
Jetzt siehst du die drei Cluster in drei verschiedenfarbigen Kästchen. Du kannst auch die Funktion color_branches() aus der Bibliothek dendextend verwenden, um deinen Baum mit verschiedenfarbigen Ästen zu visualisieren.
Denke daran, dass du ein Paket in R mit dem Befehl install.packages('package_name', dependencies = TRUE) installieren kannst.
suppressPackageStartupMessages(library(dendextend))
avg_dend_obj <- as.dendrogram(hclust_avg)
avg_col_dend <- color_branches(avg_dend_obj, h = 3)
plot(avg_col_dend)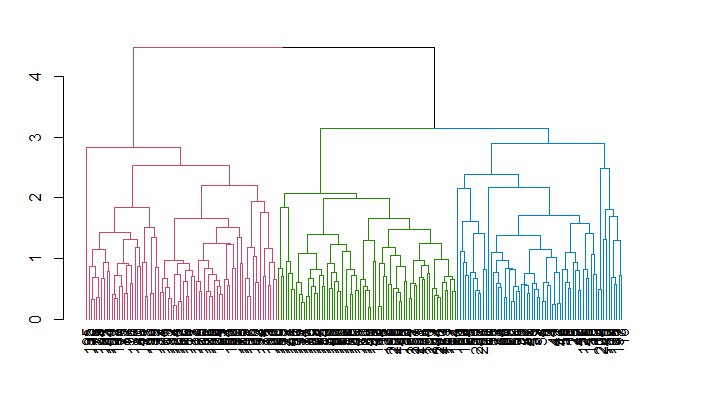
Nun fügst du die erhaltenen Clusterergebnisse wieder in den ursprünglichen DataFrame unter dem Spaltennamen cluster mit mutate() aus dem Paket dplyr ein und zählst mit der Funktion count(), wie viele Beobachtungen den einzelnen Clustern zugeordnet wurden.
suppressPackageStartupMessages(library(dplyr))
seeds_df_cl <- mutate(seeds_df, cluster = cut_avg)
count(seeds_df_cl,cluster)Du kannst sehen, wie viele Beobachtungen in jedem Cluster zugeordnet wurden. Beachte, dass du anhand der beschrifteten Daten in Wirklichkeit 70 Beobachtungen für jede Weizensorte hattest.
Es ist üblich, den Trend zwischen zwei Merkmalen auf der Grundlage des von dir durchgeführten Clustering zu bewerten, um mehr nützliche Erkenntnisse aus den Daten zu gewinnen. Als Übung kannst du mit Hilfe des Pakets ggplot2 den Trend zwischen Weizen perimeter und area clustermäßig analysieren.
suppressPackageStartupMessages(library(ggplot2))
ggplot(seeds_df_cl, aes(x=area, y = perimeter, color = factor(cluster))) + geom_point()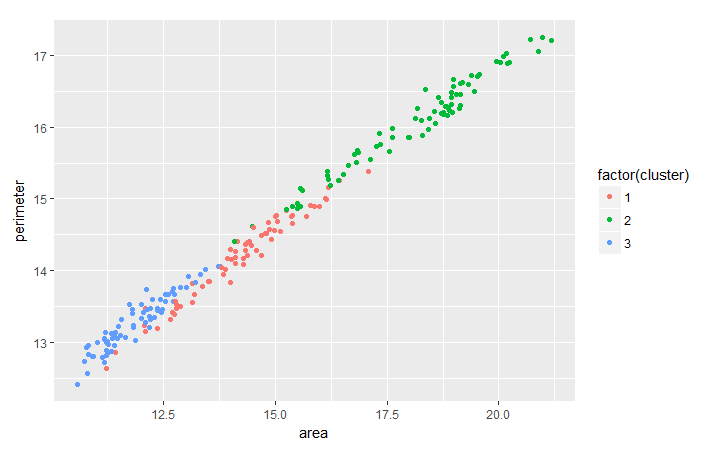
Beachte, dass es für alle Weizensorten eine lineare Beziehung zwischen ihrem Umfang und ihrer Fläche zu geben scheint.
Da du bereits die wahren Bezeichnungen für diesen Datensatz hast, kannst du deine Clustering-Ergebnisse auch mit der Funktion table() überprüfen.
table(seeds_df_cl$cluster,seeds_label)Wenn du einen Blick auf die Tabelle wirfst, die erstellt wurde, siehst du drei Gruppen mit 55 oder mehr Elementen. Insgesamt kannst du sagen, dass deine Cluster die verschiedenen Saatgutarten angemessen repräsentieren, denn ursprünglich hattest du 70 Beobachtungen für jede Weizensorte. Die größeren Gruppen stellen die Übereinstimmung zwischen den Clustern und den tatsächlichen Typen dar.
Beachte, dass du in vielen Fällen gar nicht die richtigen Etiketten hast. In diesen Fällen kannst du, wie bereits erwähnt, andere Maßnahmen ergreifen, wie z.B. die Maximierung des Dunn'schen Indexes. Du kannst den Dunn'schen Index berechnen, indem du die Funktion dunn() aus der Bibliothek clValid verwendest. Du kannst auch eine Kreuzvalidierung der Ergebnisse in Erwägung ziehen, indem du wie bei jedem anderen Algorithmus für maschinelles Lernen eine Trainings- und eine Testmenge erstellst und dann das Clustering durchführst, wenn du die echten Labels hast.
Vielleicht hast du schon vom k-means Clustering Algorithmus gehört; wenn nicht, schau dir dieses Tutorial an. Es gibt viele grundlegende Unterschiede zwischen den beiden Algorithmen, obwohl jeder von ihnen in verschiedenen Fällen besser abschneiden kann als der andere. Einige der Unterschiede sind:
Herzlichen Glückwunsch! Du hast es bis zum Ende dieses Tutorials geschafft. Du hast gelernt, wie du deine Daten vorverarbeitest, die Grundlagen des hierarchischen Clustering und die Distanzmetriken und Verknüpfungsmethoden, mit denen es arbeitet, sowie seine Verwendung in R. Du weißt auch, wie sich das hierarchische Clustering vom k-means-Algorithmus unterscheidet. Gut gemacht! Aber es gibt immer noch viel mehr zu lernen. Ich schlage vor, du schaust dir unseren Kurs "Unüberwachtes Lernen in R " an.
Weiter lernen
Kurs
Kurs
Kurs