Cours
Apprentissage non supervisé en Python
4 h
181.4K
Le SVM est un algorithme passionnant dont les concepts sont relativement simples. Le classificateur sépare les points de données à l'aide d'un hyperplan ayant la plus grande marge. C'est pourquoi un classificateur SVM est également connu sous le nom de classificateur discriminant. Le SVM trouve un hyperplan optimal qui aide à classer les nouveaux points de données.
Dans ce tutoriel, vous allez couvrir les sujets suivants :
Regardez et apprenez-en plus sur les machines à vecteurs de support avec Scikit-learn dans cette vidéo de notre cours.
Généralement, les machines à vecteurs de support sont considérées comme une approche de classification, mais elles peuvent être employées dans les deux types de problèmes de classification et de régression. Il peut facilement traiter plusieurs variables continues et catégorielles. Les SVM construisent un hyperplan dans un espace multidimensionnel pour séparer les différentes classes. Le SVM génère un hyperplan optimal de manière itérative, qui est utilisé pour minimiser l'erreur. L'idée centrale des SVM est de trouver un hyperplan marginal maximal (MMH) qui divise au mieux l'ensemble de données en classes.
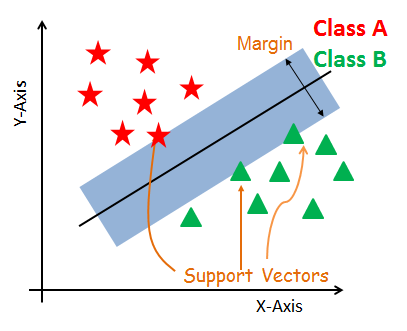
Les vecteurs de soutien sont les points de données les plus proches de l'hyperplan. Ces points permettront de mieux définir la ligne de séparation en calculant les marges. Ces points sont plus pertinents pour la construction du classificateur.
Un hyperplan est un plan de décision qui sépare un ensemble d'objets appartenant à des classes différentes.
Une marge est un espace entre les deux lignes des points de classe les plus proches. Il s'agit de la distance perpendiculaire entre la ligne et les vecteurs de support ou les points les plus proches. Si la marge est plus grande entre les classes, elle est considérée comme une bonne marge, une marge plus petite est considérée comme une mauvaise marge.
L'objectif principal est de séparer l'ensemble de données donné de la meilleure façon possible. La distance entre les deux points les plus proches est appelée marge. L'objectif est de sélectionner un hyperplan avec la plus grande marge possible entre les vecteurs de support dans l'ensemble de données donné. Le SVM recherche l'hyperplan marginal maximal dans les étapes suivantes :
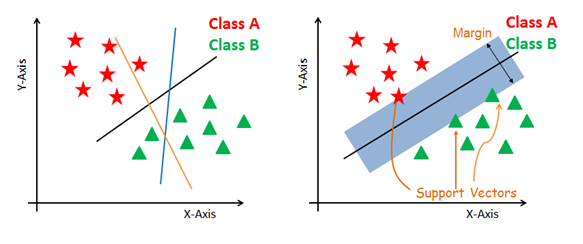
Générer des hyperplans qui séparent les classes de la meilleure façon. Figure de gauche montrant trois hyperplans noir, bleu et orange. Ici, le bleu et l'orange ont une erreur de classification plus élevée, mais le noir sépare correctement les deux classes.
Sélectionnez l'hyperplan de droite avec la ségrégation maximale à partir des deux points de données les plus proches, comme le montre la figure de droite.
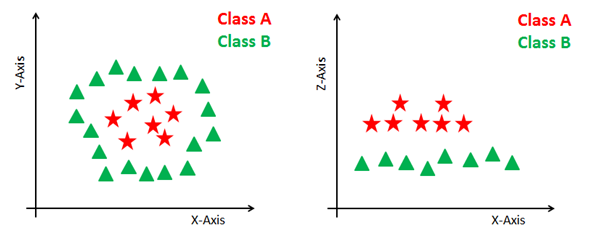
Certains problèmes ne peuvent pas être résolus à l'aide d'un hyperplan linéaire, comme le montre la figure ci-dessous (partie gauche).
Dans ce cas, le SVM utilise un noyau pour transformer l'espace d'entrée en un espace de dimension supérieure, comme indiqué à droite. Les points de données sont représentés sur l'axe des x et l'axe des z (Z est la somme quadratique de x et y : z=x^2=y^2). Vous pouvez maintenant facilement séparer ces points à l'aide de la séparation linéaire.
L'algorithme SVM est mis en œuvre dans la pratique à l'aide d'un noyau. Un noyau transforme un espace de données d'entrée dans la forme requise. Les SVM utilisent une technique appelée "astuce du noyau". Ici, le noyau prend un espace d'entrée de faible dimension et le transforme en un espace de dimension supérieure. En d'autres termes, on peut dire qu'il convertit des problèmes non séparables en problèmes séparables en leur ajoutant une dimension supplémentaire. Il est particulièrement utile pour les problèmes de séparation non linéaires. L'astuce du noyau vous permet de construire un classificateur plus précis.
K(x, xi) = sum(x * xi)
K(x,xi) = 1 + sum(x * xi)^d
Où d est le degré du polynôme. d=1 est similaire à la transformation linéaire. Le degré doit être spécifié manuellement dans l'algorithme d'apprentissage.
K(x,xi) = exp(-gamma * sum((x – xi^2))
Ici, gamma est un paramètre compris entre 0 et 1. Une valeur plus élevée de gamma correspondra parfaitement à l'ensemble des données d'apprentissage, ce qui entraînera un surajustement. Gamma=0,1 est considéré comme une bonne valeur par défaut. La valeur de gamma doit être spécifiée manuellement dans l'algorithme d'apprentissage.
Jusqu'à présent, vous avez appris les fondements théoriques des SVM. Vous allez maintenant découvrir sa mise en œuvre en Python à l'aide de scikit-learn.
Pour la construction du modèle, vous pouvez utiliser l'ensemble de données sur le cancer, qui est un problème de classification multi-classes très connu. Cet ensemble de données est calculé à partir d'une image numérisée d'une aspiration à l'aiguille fine (AAF) d'une masse mammaire. Ils décrivent les caractéristiques des noyaux cellulaires présents dans l'image.
L'ensemble de données comprend 30 caractéristiques (rayon moyen, texture moyenne, périmètre moyen, surface moyenne, douceur moyenne, compacité moyenne, concavité moyenne, points concaves moyens, symétrie moyenne, dimension fractale moyenne, erreur de rayon, erreur de texture, erreur de périmètre, erreur de surface, erreur de douceur, erreur de compacité, erreur de symétrie, erreur de dimension fractale, pire rayon, pire texture, pire périmètre, pire surface, pire douceur, pire compacité, pire concavité, pire points concaves, pire dimension fractale), erreur de concavité, erreur de points concaves, erreur de symétrie, erreur de dimension fractale, pire rayon, pire texture, pire périmètre, pire surface, pire lissage, pire compacité, pire concavité, pire points concaves, pire symétrie et pire dimension fractale) et une cible (type de cancer).
Ces données comportent deux types de classes de cancer : les cancers malins (nocifs) et les cancers bénins (non nocifs). Ici, vous pouvez construire un modèle pour classer le type de cancer. L'ensemble de données est disponible dans la bibliothèque scikit-learn ou vous pouvez également le télécharger à partir de la bibliothèque d'apprentissage automatique de l'UCI.
Exécutez et modifiez le code de ce tutoriel en ligne
Exécuter le codeCours Scikit-learn
Cours
Cours
blog
Nathaniel Taylor-Leach
8 min
blog
Nisha Arya Ahmed
15 min
blog
Nathaniel Taylor-Leach
blog
Nathaniel Taylor-Leach
blog
Fereshteh Forough
4 min